HiFi-MAG-Pipeline
Overview
The purpose of this snakemake workflow is to obtain high-quality metagenome-assembled genomes (MAGs) from previously generated assemblies. This workflow received major improvements in v2.0 (Feb 2023). The new steps of the HiFi-MAG-Pipeline are shown below:
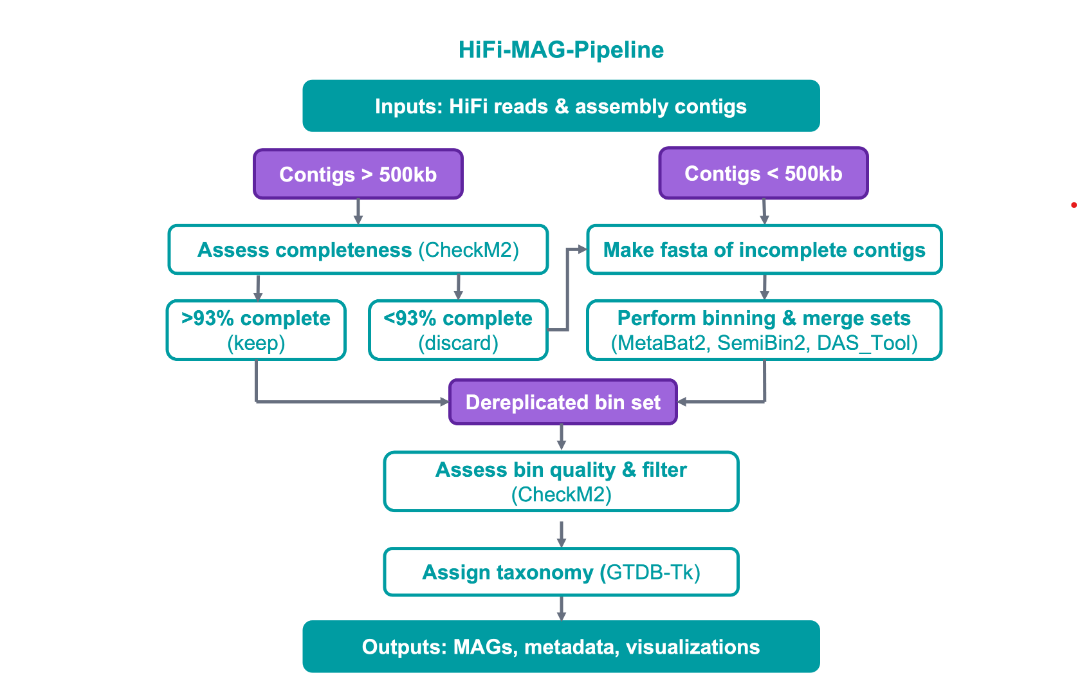
The new version of this workflow is "completeness-aware". Long contigs > 500 kb are identified and placed in individual fasta files. They are then examined using CheckM2 to determine percent completeness. All long contigs that are > 93 % complete are then moved directly to the final MAG set.
The long contigs that are > 93% complete are pooled with other shorter incomplete contigs from the starting set, and this contig set is subjected to binning. Binning algorithms include MetaBat2 and SemiBin2 (using long read settings). The two bin sets are compared and merged using DAS_Tool.
The dereplicated bin set consists of the merged bin set from above and all long complete contigs found. The dereplicated bin set is examined using CheckM2, and subsequently filtered based on several qualities (defaults >= 70% completeness, > 10% contamination, > 20 contigs).
All bins/MAGs passing filtering undergo taxonomic assignment using GTDB-Tk. The final MAGs are written as a set of fasta files, several figures are produced, and a summary file of metadata is generated.